






药物筛选与虚拟筛选(Schrödinger/GROMACS/AMBER):工作站与服务器配置全解析
时间:2026-02-25 23:00:57
来源:UltraLAB图形工作站方案网站
人气:3261
作者:管理员
——从百万分子库虚拟筛选到微秒级分子动力学轨迹,从结合自由能计算到AI生成式药物设计,算力如何加速新药发现的"最后一公里"?
引言:当药物发现进入"算力密集型"时代
在传统药物研发平均耗时10年、耗资26亿美元的沉重背景下,计算机辅助药物设计(CADD)与AI制药(AIDD)正成为破局的关键。从早期基于分子对接(Molecular Docking)的虚拟筛选(Virtual Screening, VS),到精细化的分子动力学(Molecular Dynamics, MD)模拟,再到高精度的自由能微扰(Free Energy Perturbation, FEP)计算,以及近年来大语言模型(LLM)驱动的分子生成与性质预测,计算生物学正经历着从"辅助验证"到"主导发现"的范式转移。
然而,当面对数千万分子的虚拟筛选库、百万原子级别的膜蛋白-配体复合体、或是需要数微秒甚至毫秒级轨迹的别构效应(Allosteric Effect)研究时,普通的办公电脑甚至传统的生命科学计算集群都会暴露出严重的性能瓶颈:Schrödinger Glide对接任务在单核CPU上排队到天荒地老,GROMACS模拟因内存不足无法加载全原子膜蛋白体系,AMBER的FEP计算因显存溢出而中断,庞大的轨迹文件(Trajectory)让机械硬盘读写灯彻夜狂闪导致系统假死...
作为深耕科学计算硬件领域的UltraLAB,我们在服务恒瑞医药、药明康德、中科院上海药物所、北大药学院等顶级药物研发机构的过程中,深刻认识到:药物筛选计算是"高吞吐、高并发、高IO"的典型代表,其硬件配置需要精准匹配不同计算阶段的算法特征。本文将深度解构Schrödinger、GROMACS、AMBER等主流软件的硬件需求,并提供从桌面工作站到GPU集群的UltraLAB全栈解决方案。
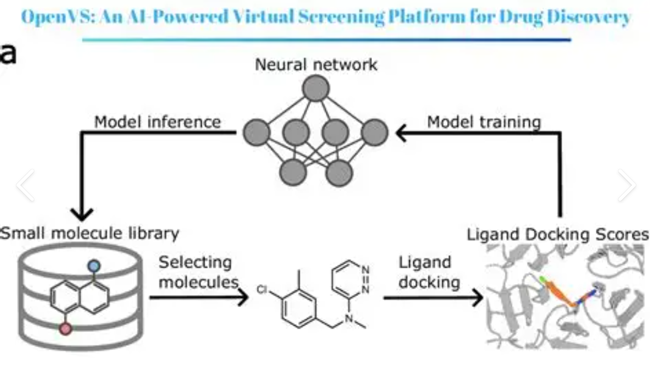
一、药物筛选全链条:计算类型与硬件需求映射
1.1 虚拟筛选(Virtual Screening):高吞吐 embarrassingly parallel
计算特征:
-
分子对接(Docking):Schrödinger Glide、AutoDock Vina、OpenEye等,评估配体-受体结合构象与亲和力;
-
任务并行度:数百万分子可完全独立计算,embarrassingly parallel,几乎线性加速;
-
计算密度:每个对接任务计算量中等,但需处理大量构象搜索(Conformational Search);
-
精度梯度:从高通量虚拟筛选(HTVS)到标准精度(SP)再到高精度(XP),计算成本指数增长。
硬件瓶颈:
-
CPU核心数:单分子对接通常单线程或轻度多线程(<4核),高并发是关键,需要"多核+高主频"平衡;
-
内存:单个对接任务占用内存不大(<2GB),但并发数百任务时,总内存需求可达数百GB;
-
存储I/O:数百万小文件(输入/输出分子结构)的随机读写,对传统HDD是灾难。
UltraLAB配置策略:
-
高频多核CPU:AMD EPYC 9654(96核)或Intel Xeon W-3400系列,支持同时运行96-128个并行对接任务;
-
大内存:256GB-512GB DDR4/DDR5 ECC,支持高并发时的内存冗余;
-
高速存储:NVMe SSD阵列(RAID 0),应对海量小文件的并发读写。
1.2 分子动力学(MD):GPU加速的"显存墙"困境
计算特征:
-
力场计算:每时间步计算键合相互作用(Bonded)与非键合相互作用(Non-bonded,静电与范德华);
-
长程静电:PME(Particle Mesh Ewald)方法将计算复杂度从O(N2) 降至O(NlogN) ,但需三维FFT;
-
时间尺度困境:典型时间步长2fs,模拟1微秒需5亿步,总计算量巨大;
-
体系规模:全原子膜蛋白(如GPCR、离子通道)轻松超过10万原子,含显式水分子可达百万原子。
硬件瓶颈:
-
GPU显存:AMBER的PMEMD.cuda或GROMACS的GPU加速,需将整个体系坐标、力场参数驻留显存;
-
10万原子体系:显存占用~2-4GB;
-
百万原子体系:显存占用>20GB,需RTX A6000 48GB或A100 80GB;
-
-
双精度需求:NVT/NPT系综的约束算法(SHAKE/SETTLE/LINCS)对数值稳定性敏感,双精度(FP64)更稳定,消费级RTX显卡(FP64性能阉割)可能积累误差;
-
存储带宽:微秒级轨迹文件可达数TB(每帧数MB × 数十万帧),写入速度瓶颈明显。
1.3 自由能计算(FEP/TI):精算密集型任务
计算特征:
-
炼金术变换(Alchemical Transformation):逐步"消失"一个配体同时"出现"另一个,计算结合自由能差ΔΔG ;
-
Schrödinger FEP+:工业金标准,采用Replica Exchange with Solute Tempering (REST) 增强采样;
-
AMBER TI/TI-4PB:热力学积分计算自由能;
-
收敛要求:需数十至数百个λ 窗口,每个窗口数ns至数十ns模拟,总计算量是普通MD的10-100倍。
硬件需求:
-
GPU性能:FEP+强烈推荐NVIDIA Tesla/Quadro/RTX专业卡,利用Tensor Core加速;
-
多副本并行:不同λ 窗口可完全并行,适合多GPU配置;
-
通信延迟:副本交换(Replica Exchange)需频繁交换坐标/能量信息,对多节点集群的网络延迟敏感(需InfiniBand)。
二、主流软件硬件特性深度解析
2.1 Schrödinger套件:商业软件的GPU饥渴症
Glide(对接):
-
CPU并行:支持多线程,但扩展性有限(通常<8核/任务);
-
多任务并发:适合单机多核并行处理大量配体。
Desmond(MD):
-
GPU加速:Desmond-GPU版本对NVIDIA GPU优化极佳,百万原子体系在A100上可达>100 ns/day;
-
显存需求:Desmond的显存管理高效,但大体系仍需>24GB显存。
FEP+(自由能):
-
硬件门槛:必须使用NVIDIA GPU(Volta架构及以上,即V100/A100/RTX 30/40系列),且需双精度支持;
-
多GPU扩展:支持单节点多GPU并行(Replica Exchange),多节点扩展需高速网络;
-
许可限制:Schrödinger许可与硬件绑定,需考虑License Server的稳定性(建议双机热备)。
UltraLAB优化:
-
配置4× RTX A6000 48GB或8× A100 80GB NVLink服务器,满足FEP+的多副本并行需求;
-
安装Schrödinger优化版Linux内核,减少系统抖动对GPU计算的影响。
2.2 GROMACS:开源利器与异构计算典范
算法优势:
-
领域分解(Domain Decomposition):将体系空间划分到不同MPI rank,适合大规模集群;
-
负载均衡:动态负载均衡(Dynamic Load Balancing)适应非均相体系(如膜蛋白-水混合);
-
GPU卸载:可将非键合计算(PME与短程力)完全卸载至GPU,CPU仅负责约束与积分。
硬件配置黄金法则:
-
CPU-GPU配比:推荐1-2 CPU核心 : 1 GPU的比例,避免CPU成为瓶颈;
-
内存带宽:GROMACS对内存带宽敏感,八通道DDR5(>400GB/s)显著优于四通道;
-
PME网格:长程静电计算的3D-FFT在GPU上效率极高,但需足够显存存储网格。
典型配置:
-
单机:AMD Threadripper PRO 5995WX(64核)+ 4× RTX A5000(16GB),平衡成本与性能;
-
集群:双路AMD EPYC 9654(96核×2)+ 8× A100 80GB,通过MPI实现数百ns/day的吞吐。
2.3 AMBER:经典力场与高性能CUDA
PMEMD.cuda:
-
精度要求:虽支持单精度(SPFP)和混合精度(DPFP),但长程静电与约束求解建议双精度以确保能量守恒;
-
显存优化:相比GROMACS,AMBER对显存的需求略低,但大体系(>50万原子)仍需24GB+;
-
多GPU支持:AMBER 22+支持多GPU并行(PMEMD.cuda.MPI),但扩展效率随GPU数增加而下降(通常<4卡效率较高)。
硬件建议:
-
对于常规MD:RTX 4090(24GB)性价比极高,但需注意散热与稳定性;
-
对于FEP/TI:建议使用A100或RTX A6000,确保数值稳定性与ECC纠错。
三、UltraLAB药物筛选算力配置体系
针对药物发现的不同阶段,UltraLAB提供差异化硬件方案:
方案A:虚拟筛选专用工作站(Hit Identification)
目标:处理百万级分子库的高通量对接(Glide HTVS/SP)
配置:UltraLAB VS-Station Pro
-
CPU:AMD EPYC 9554(64核,3.1GHz Base),支持同时运行64个对接任务;
-
内存:512GB DDR5-4800 ECC REG(8×64GB),支持高并发内存需求;
-
存储:
-
系统盘:2TB NVMe Gen4(存放分子库与软件);
-
数据盘:8TB NVMe U.2企业级RAID 0(顺序读写>12GB/s,应对数百万分子的并行I/O);
-
-
网络:双端口10GbE,支持从公司数据库高速下载化合物库;
-
预估性能:单日可完成50-100万分子的Glide SP对接筛选。
方案B:分子动力学模拟服务器(Lead Optimization)
目标:膜蛋白体系(GPCR、激酶)的百纳秒至微秒级模拟,FEP计算
配置:UltraLAB MD-GPU Server
-
GPU:8× NVIDIA RTX A6000 48GB(NVLink桥接,显存池化可达384GB)
-
支持AMBER/GROMACS的多GPU并行;
-
48GB显存可承载200万+原子的体系(全原子膜蛋白+水+离子);
-
-
CPU:2× Intel Xeon Gold 6458Q(32核×2,共64核,睿频3.4GHz)
-
匹配8 GPU的CPU需求,避免CPU瓶颈;
-
-
内存:1TB DDR5-4800 ECC(16×64GB)
-
预加载大体系拓扑文件,支持轨迹实时分析;
-
-
存储:RAID 0 NVMe阵列(4×7.68TB),持续写入速度>20GB/s,确保微秒级轨迹(数TB)快速落盘;
-
散热:工业级风道设计,支持GPU 7×24小时满载运行不降频。
性能基准:
-
GROMACS:百万原子膜蛋白体系,A6000单卡可达80-100 ns/day;
-
Schrödinger FEP+:8卡并行可同时处理8个配体对的FEP计算,或单个配体的8副本REST模拟。
方案C:AI制药与生成式模型训练(AIDD)
目标:AlphaFold结构预测、Diffusion模型分子生成、大规模图神经网络(GNN)训练
配置:UltraLAB AI-Pharma Cluster
-
GPU计算节点:4× 节点,每节点8× NVIDIA A100 80GB(NVLink + NVSwitch互联)
-
支持AlphaFold-Multimer的大蛋白复合体预测;
-
支持MegaMolBART、ChemBERTa等大模型的分布式训练;
-
-
CPU:每节点2× AMD EPYC 9654(96核×2),负责数据预处理与特征工程;
-
内存:每节点2TB DDR5,支持大分子结构的内存驻留;
-
存储:并行文件系统(BeeGFS),聚合带宽>100GB/s,存储容量>500TB,支持数百万蛋白质结构文件的随机访问;
-
网络:InfiniBand HDR(200Gbps),确保多节点GPU间的梯度同步延迟<1μs。
方案D:综合药物发现平台(一站式解决方案)
适用:CRO公司或大型药企计算化学部门
混合架构:
-
登录/调度节点:UltraLAB EX650i(管理作业提交、License管理、数据归档);
-
虚拟筛选队列:4× CPU胖节点(每节点128核,1TB内存),专责对接计算;
-
MD/FEP队列:8× GPU节点(每节点4× A100),专责分子动力学;
-
AI训练队列:2× AI训练节点(8× A100),专责模型训练;
-
存储层:
-
热数据:全闪存阵列(>50TB NVMe),存放当前项目轨迹;
-
温数据:HDD阵列(>500TB),存放历史项目数据;
-
冷数据:对象存储/磁带库,合规存档。
-
四、关键硬件选型技术细节
4.1 GPU选型:游戏卡 vs 专业卡 vs 计算卡
| 型号 | 显存 | FP64性能 | ECC | 适用场景 | UltraLAB建议 |
|---|---|---|---|---|---|
| RTX 4090 | 24GB GDDR6X | 1/64 FP32 | 无 | 小规模MD(<50万原子)、教学使用 | 性价比之选,但需接受无ECC风险,适合虚拟筛选后的精修 |
| RTX A5000 | 16GB GDDR6 | 1/2 FP32 | 有 | 中等规模MD、FEP计算 | 均衡之选,适合预算有限的FEP研究 |
| RTX A6000 | 48GB GDDR6 | 1/2 FP32 | 有 | 大规模膜蛋白、长时程模拟 | 推荐配置,大显存可承载超大体系,ECC保障数据完整性 |
| A100 80GB | 80GB HBM2e | 1/2 FP32 | 有 | 超大规模体系、AI训练、FEP+ | 极致性能,HBM显存带宽>2TB/s,适合对速度敏感的项目 |
关键提示:
-
显存容量优先:药物筛选中,体系规模(原子数)决定显存下限,宁可降速(用A6000而非A100)也要避免显存溢出(OOM);
-
NVLink必要性:多GPU并行时,NVLink的900GB/s带宽远高于PCIe的64GB/s,对于FEP的Replica Exchange或GROMACS的PME网格交换至关重要。
4.2 存储架构:应对Trajectory洪流
分子动力学轨迹文件是存储的"黑洞":
-
100万原子体系,每100ps保存一帧,模拟1微秒(1000帧):
-
每帧大小:100万原子 × 3坐标 × 8字节(双精度)≈ 24MB;
-
总大小:24GB;
-
若每天产生1个微秒轨迹,月数据量>700GB。
-
UltraLAB存储优化:
-
分层存储策略:
-
L1(热层):NVMe SSD(>10GB/s),存放正在分析的轨迹(最近1周数据);
-
L2(温层):SAS HDD RAID 6(>1GB/s),存放已完成项目的原始轨迹;
-
L3(冷层):LTO-9磁带库(18TB/盘),长期存档(符合FDA 21 CFR Part 11等合规要求)。
-
-
轨迹压缩:配置MDAnalysis/VMD后端,自动压缩轨迹(如XTC格式替代TRR),节省70%空间;
-
内存分析:对于超大轨迹(>100GB),使用Memory-Mapped I/O技术,允许程序直接访问存储而不全量加载至内存。
4.3 网络与数据管理
虚拟筛选的数据流动:
-
化合物库(数百万SDF/MOL2文件)通常存储于企业NAS;
-
计算节点需高速读取(避免成为瓶颈),建议10GbE/25GbE网络,或直接将热数据缓存于本地NVMe。
集群并行通信:
-
GROMACS多节点:使用MPI+OpenMP混合并行,Domain Decomposition跨节点时需频繁交换ghost原子坐标;
-
网络要求:InfiniBand HDR(100Gbps+,延迟<1μs),确保弱扩展效率(Weak Scaling)>80%。
五、软件优化与硬件协同
5.1 Schrödinger套件优化
-
FEP+的GPU选择:确保驱动版本≥510.x,CUDA≥11.6,启用Persistent Mode减少GPU初始化延迟;
-
Job Control:利用Schrödinger Job Control Center,将任务自动分发至多节点GPU队列;
-
License优化:配置本地License Server,避免网络波动导致的计算中断。
5.2 GROMACS性能调优
-
平衡CPU-GPU负载:使用
gmx mdrun -nb gpu -pme gpu -bonded cpu,将键合作用保留在CPU,非键合卸载至GPU,避免CPU瓶颈; -
压缩轨迹输出:使用
-x选项输出压缩XTC,减少IO压力; -
多副本并行(Replica Exchange):利用
gmx mdrun -multidir启动多个独立模拟,最大化GPU利用率。
5.3 AMBER配置
-
显存优化:使用
pmemd.cuda_DPFP(双精度)确保能量守恒,或pmemd.cuda_SPFP(单精度)提升速度(需验证体系稳定性); -
多GPU设置:设置
CUDA_VISIBLE_DEVICES环境变量,确保每个AMBER实例绑定特定GPU,避免资源争抢。
结语:以算力为基,筑新药发现之塔
从AlphaFold破解蛋白质折叠之谜,到AI生成式模型设计出自然界不存在的分子骨架,从虚拟筛选中"大海捞针"发现先导化合物,到FEP计算精确预测结合亲和力以替代部分湿实验,计算生物学正站在一个前所未有的算力拐点。然而,这些突破的背后,是分子动力学模拟对GPU显存的贪婪渴求,是虚拟筛选对CPU并发能力的极致压榨,是自由能计算对数值精度的严苛要求。
UltraLAB深刻理解药物研发"时间就是生命"的紧迫性。我们提供的不仅是堆砌核心与显卡的硬件,而是针对Schrödinger、GROMACS、AMBER等软件特性深度优化的算力解决方案:从确保FEP+数值稳定性的ECC显存配置,到应对百万分子I/O风暴的NVMe存储架构,再到支持多副本并行的高带宽网络,每一处细节都旨在让您的模拟跑得更快、更稳、更准。
无论是初创Biotech公司的第一台MD工作站,还是大型Pharma的智能化药物发现平台,UltraLAB都能提供从桌面到数据中心的全谱系支持。在对抗疾病的漫长征途中,让我们用坚实的算力基础设施,为您的创新分子设计加速——因为每一个提前计算出的有效结合模式,都可能意味着患者提前获得救治的希望。
关于UltraLAB生命科学计算解决方案
UltraLAB专注于药物发现与计算生物学高性能计算系统的定制,产品覆盖虚拟筛选、分子动力学、自由能计算、AI制药等全场景。我们提供符合GMP/GxP规范的硬件可靠性设计,支持Schrödinger、GROMACS、AMBER、AlphaFold、OpenEye等主流软件的优化部署,并提供从硬件选型、集群搭建到软件调优的一站式服务。


