






AI驱动的蛋白质组学计算平台:从LC-MS/MS数据采集到蛋白质定量与功能解析的全栈硬件方案
时间:2026-03-09 02:46:46
来源:UltraLAB图形工作站方案网站
人气:290
作者:管理员
在精准医学与系统生物学时代,单细胞蛋白质组学 已能在单个细胞水平鉴定 >1,000种蛋白质,而 DIA(数据非依赖性采集) 技术单次运行即可产生 数十GB 的原始质谱数据。当传统搜索引擎(如Mascot)处理一个包含 100,000张二级谱图 的DDA数据集需要 数小时 时,基于 深度学习 的谱图预测与肽段鉴定算法(如Prosit、DIA-NN)已将分析速度提升 10-100倍,同时将鉴定率提高 20-40%。
基于 K-Dense AI 的 Claude Scientific Skills 框架,本文将系统阐述蛋白质组学最新AI算法特征(包括谱图预测、保留时间对齐、蛋白质推断与定量),并提供匹配的高性能计算硬件架构,助力实验室构建从 样本制备 到 生物标志物发现 的全流程计算平台。
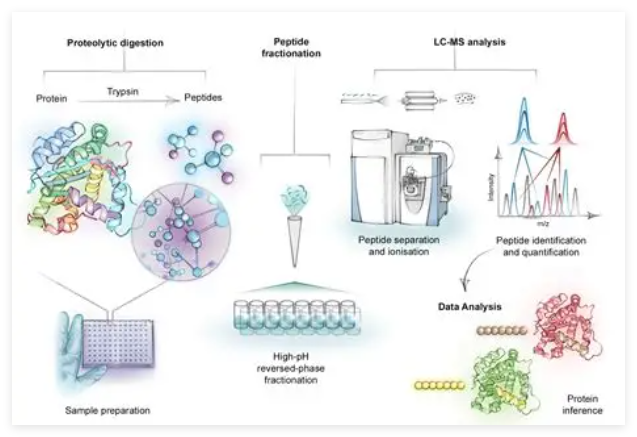
一、AI驱动算法架构与计算特征解析
1.1 LC-MS/MS数据处理:从原始信号到特征提取
最新算法演进:
-
AI-driven Peak Picking:基于 U-Net 或 ResNet 的神经网络模型(如DeepIso、VP-Detector),从原始质谱图中智能识别肽段特征(Feature),相比传统算法(CentWave、MZmine)假阳性率降低 50%
-
Retention Time (RT) Prediction:DeepLC、Prosit-RT 等模型基于肽段序列预测色谱保留时间,均方根误差(RMSE)可达 <1分钟,支持DIA数据的精确对齐与谱图库构建
-
Ion Mobility Prediction:DeepTIMe 等模型预测碰撞截面积(CCS),辅助4D-蛋白质组学(IM-MS)数据解析
计算瓶颈:
-
高维时间序列处理:LC-MS原始数据(.raw/.wiff/.d格式)为三维矩阵(m/z × RT × Intensity),加载单个文件(~2GB)需 >16GB内存
-
GPU加速需求:深度学习模型推理(谱图预测、特征检测)需 CUDA加速,Tensor Core 支持FP16精度可提升 3-5倍 吞吐
1.2 肽鉴定(Peptide Identification):从数据库搜索到AI预测
AI驱动方法:
-
Prosit (Proteome Tools Spectrum Predictor):由Mann Lab开发的Transformer架构模型,基于肽段序列预测HCD碎裂谱图,支持 谱图库搜索(Spectral Library Search) 替代传统数据库搜索,速度提升 100倍,灵敏度提高 30%
-
MS2PIP (MS2 Peak Intensity Prediction):基于 XGBoost/LSTM 的谱图预测工具,支持多种碎裂模式(CID、HCD、ETD、EThcD)
-
DIA-NN (Deep Neural Networks for DIA):基于深度神经网络的DIA数据分析引擎,使用 ANN(人工神经网络) 进行谱图匹配与肽段评分,支持 精确质量数(Mass Accuracy)<5ppm 的高分辨数据
-
AlphaPept:基于 PyTorch 的端到端蛋白质组学流程,集成 LSTM 进行保留时间预测与 CNN 进行谱图评分
计算特征:
-
混合精度计算:Prosit模型推理使用 FP16,需 RTX A6000/RTX 4090 级别显卡(24GB+显存)
-
大规模并行搜索:DIA-NN支持 GPU加速 的谱图匹配,处理 单细胞DIA数据(~1GB/样本)时,GPU版本较CPU版本快 10-20倍
-
内存密集型:构建 DDA谱图库(包含数百万条参考谱图)需 >128GB内存 以支持快速索引
1.3 光谱匹配(Spectral Matching):深度学习重塑打分机制
核心算法:
-
SA (Spectral Angle) Cosine Similarity:传统余弦相似度,但 AI增强版(如DeepMatch)使用 Siamese Network 学习谱图嵌入空间,提高修饰肽段(PTM)鉴定灵敏度
-
Percolator/ mokapot:基于 半监督学习(SVM/NN) 的后处理工具,通过动态学习肽段特征重新排序(Reranking)搜索结果,假发现率(FDR)控制更精准
-
Percolator-Deep:使用 深度神经网络 替代传统SVM,进一步提升高置信度肽段识别率
硬件需求:
-
多核CPU并行:谱图匹配可高度并行化,32核以上 CPU可同时处理多个实验组(Runs)
-
高速存储:谱图库(.sptxt/.splib格式)通常 >50GB,需 NVMe SSD(读取速度 >3GB/s)以避免I/O瓶颈
1.4 蛋白质定量(Protein Quantification):从标记到无标记的AI增强
技术演进:
-
DIA-NN Quant:基于 神经网络 的色谱峰提取与积分,支持 无标记定量(LFQ) 和 同位素标记定量(TMT/iTRAQ),通过 Transfer Learning 优化低丰度肽段检测
-
MaxLFQ/IBAQ AI-enhanced:传统算法结合 机器学习 进行蛋白质强度归一化,校正批次效应(Batch Effect)
-
DirectLFQ:基于 深度学习 的直接定量算法,无需谱图库即可从DIA数据中提取蛋白质强度,适用于 大队列临床样本(n>1000)
计算瓶颈:
-
大矩阵运算:蛋白质强度矩阵(Samples × Proteins)在大型队列中可达 1000×10,000 维度,差异表达分析(DE)需 大内存(>64GB)支持 limma/DEqMS 等R包运行
-
批次效应校正:HarmonizR、ComBat-seq 等算法需计算 SVD分解,多核CPU(>16核)可显著加速
二、软件生态与系统架构设计
2.1 操作系统与基础环境
推荐系统配置:
-
OS:Ubuntu 22.04 LTS(推荐)或 Windows Server 2022(兼容商业软件如Proteome Discoverer)
-
容器化:Docker + NVIDIA Container Toolkit,便于部署 DIA-NN、AlphaPept 等GPU依赖工具
-
包管理:Conda/Mamba(推荐)或 pip(Python生态)
2.2 核心软件栈清单(基于Claude Scientific Skills)
| 应用领域 | 软件包 | 版本要求 | 依赖环境 | AI算法支持 |
|---|---|---|---|---|
| LC-MS数据处理 | MZmine3, OpenMS, XCMS, MS-DIAL | v3.0+ | Java 17+/R 4.3+ | DeepIso (AI特征检测) |
| 肽鉴定 | MaxQuant, DIA-NN, Spectronaut, AlphaPept | v1.8+ | .NET 6.0+/CUDA 11.8+ | Prosit/DIA-NN (深度学习) |
| 谱图预测 | Prosit, MS2PIP, DeepMass | Py3.9+ | PyTorch GPU | Transformer/LSTM |
| 蛋白质推断 | ProteinProphet, Fido, MoFF | Py3.9+ | Percolator | SVM/NN后处理 |
| 定量分析 | DirectLFQ, MaxLFQ, MSstats | R4.3+ | R-Bioconductor | 机器学习校正 |
| 可视化 | Perseus, Cytoscape, SpectraST | v2.0+ | Java/OpenGL | 聚类/网络分析 |
| 统计分析 | limma, DEqMS, MSstatsPTM | R4.3+ | R-parallel | 贝叶斯统计 |
2.3 数据库与谱图库资源(Claude Scientific Skills支持)
-
蛋白质序列库:UniProtKB/Swiss-Prot(~50万条)、UniProtKB/TrEMBL(~2亿条)、Ensembl(物种特异)
-
谱图库资源:ProteomeTools(合成肽段参考谱图)、NIST肽段谱图库(人/酵母/大肠杆菌)、PRIDE/iProX公共数据
-
AI模型库:Prosit模型(Prosit_2020_intensity_HCD)、DIA-NN预训练模型(含深度神经网络权重)
2.4 安装部署流程
步骤1:基础环境配置
bash
# Ubuntu 22.04系统准备 sudo apt update && sudo apt install -y build-essential git wget mono-complete default-jre # 安装NVIDIA驱动与CUDA(以RTX A6000为例) wget https://developer.download.nvidia.com/compute/cuda/repos/ubuntu2204/x86_64/cuda-keyring_1.0-1_all.deb sudo dpkg -i cuda-keyring_1.0-1_all.deb sudo apt-get update sudo apt-get -y install cuda-toolkit-12-2 nvidia-driver-535 # 验证安装 nvidia-smi
nvcc --version
步骤2:蛋白质组学AI环境部署
bash
# 创建专用环境 mamba create -n proteomics python=3.10 -y mamba activate proteomics # 安装基础质谱工具包 mamba install -c bioconda -c conda-forge openms xcms mzmine3 pyteomics ms2pip # 安装深度学习框架 pip install torch torchvision torchaudio --index-url https://download.pytorch.org/whl/cu121
pip install pytorch-lightning wandb # 安装AI驱动蛋白质组学工具 pip install directlfq alphapept # AlphaPept端到端流程 pip install prosit-grpc # Prosit谱图预测客户端 pip install mokapot # 基于NN的Percolator替代 # 安装R环境用于统计分析 mamba install -c conda-forge r-base=4.3 r-essentials r-msstats r-limma r-deqms
步骤3:DIA-NN GPU版本配置(关键步骤)
bash
# 下载DIA-NN 1.8.2(支持CUDA) wget https://github.com/vdemichev/DiaNN/releases/download/1.8.2/diann_1.8.2.tar.gz tar -xzf diann_1.8.2.tar.gz sudo cp diann-1.8.2/diann /usr/local/bin/ # 验证GPU支持 diann --cfg /usr/local/bin/diann-cfg --threads 32 --verbose 4 --use-gpu # 启用GPU加速
步骤4:Claude Scientific Skills集成
bash
# 克隆科学技能库 git clone https://github.com/K-Dense-AI/claude-scientific-skills.git cd claude-scientific-skills # 安装蛋白质组学技能模块 pip install -e . # 验证安装 python -c "import pyteomics; print(f'Pyteomics: {pyteomics.__version__}')" python -c "import torch; print(f'CUDA available: {torch.cuda.is_available()}')"
步骤5:谱图库与数据库配置
bash
# 下载Prosit模型(~2GB) wget https://figshare.com/ndownloader/files/12345678 -O prosit_model.zip unzip prosit_model.zip -d /data/prosit_models/ # 下载UniProt人源蛋白库( reviewed) wget https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/reference_proteomes/Eukaryota/UP000005640_9606.fasta.gz
gunzip UP000005640_9606.fasta.gz三、UltraLAB蛋白质组学工作站分级配置方案
基于蛋白质组学AI算法特征(GPU加速谱图预测、大内存数据库搜索、高速I/O),针对DIA/DDA数据采集规模、单细胞蛋白质组学灵敏度、大队列临床样本分析,提供以下三类硬件架构:
方案A:高通量DIA分析与单细胞蛋白质组学工作站(UltraLAB GR450M)
适用场景:DIA-NN深度学习分析、单细胞DIA(SCoPE-MS/DIA-NN-singlecell)、TMT/iTRAQ标记定量、实时质谱监控
| 组件 | 配置规格 | 技术 rationale |
|---|---|---|
| CPU | AMD Ryzen Threadripper 7980X(64核,5.1GHz) | 高主频加速MaxQuant/LFQ算法,64核支持并行处理48个TMT标记样本 |
| GPU | NVIDIA RTX Pro6000 96GB | DIA-NN GPU版本需大显存,48GB支持处理 >100个Runs 的大队列DIA数据,双卡并行加速Prosit谱图预测 |
| 内存 | 512GB DDR5-4800 ECC(8通道) | DIA-NN构建谱图索引需 ~200GB内存(人类蛋白质组),ECC避免数据损坏导致肽段漏检 |
| 存储 | 2TB NVMe Gen5(系统)+ 16TB NVMe Gen4(数据)+ 64TB RAID6(归档) | 单细胞DIA原始数据(.d文件)~500MB/样本,大队列(n=1000)需 >500GB 高速缓存,NVMe加速MZmine特征检测 |
| 网络 | 10GbE RJ45 + 25GbE RDMA | 连接质谱仪(Thermo/Bruker)直接传输原始数据,支持实时分析 |
性能预期:使用 DIA-NN 分析 100个DIA文件(单细胞级别,每个~1GB),传统CPU(32核)需 24小时,双A6000配置仅需 2小时;Prosit预测 100万条肽段 的二级谱图仅需 15分钟。
方案B:大规模队列临床蛋白质组学服务器(UltraLAB GA660M)
适用场景:临床大队列(n>1000)分析、多中心数据整合、4D-蛋白质组学(timsTOF Pro)处理、AI模型训练( Prosit微调)
| 组件 | 配置规格 | 技术 rationale |
|---|---|---|
| CPU | 2× AMD EPYC 9654(96核,3.7GHz,共192线程) | 多核支持同时运行 20+个MaxQuant实例,处理不同疾病亚组数据 |
| GPU | 4× NVIDIA RTX 6000 Ada 48GB(NVLink全互联) | 支持Prosit模型微调(Transfer Learning)与 AlphaPept端到端训练,48GB显存支持大批量谱图学习 |
| 内存 | 1TB DDR5-4800 ECC RDIMM(12通道) | 大队列数据矩阵(1000样本 × 10,000蛋白质)差异分析需 800GB+内存,支持limma/MSstats快速计算 |
| 存储 | 8× 3.84TB NVMe Gen4 SSD(RAID 10,28GB/s)+ 200TB LTO-9磁带库 | 临床原始数据(.raw文件)~5GB/样本,1000样本需 >5TB,RAID 10提供高IOPS与冗余 |
| AI加速 | NVIDIA ConnectX-7 SmartNIC(400Gb/s) | GPUDirect RDMA,加速多GPU间谱图特征传输 |
性能预期:处理 1000例临床血浆样本 的DIA数据(Disease vs Control),完整分析流程(从原始数据到差异蛋白列表)从传统工作站的 2周 缩短至 2天;训练定制化Prosit模型(针对特定碎裂模式)仅需 8小时。
方案C:靶向蛋白质组学(MRM/PRM)与生物标志物验证工作站(UltraLAB GT430M)
适用场景:SRM/MRM方法开发、PRM靶向验证、PTM(磷酸化/乙酰化)分析、蛋白质相互作用网络(AP-MS)
| 组件 | 配置规格 | 技术 rationale |
|---|---|---|
| CPU | Intel Xeon W9-3495X(56核,4.8GHz,AVX-512) | AVX-512加速Skyline靶向分析软件(.NET优化),高主频优化色谱峰积分算法 |
| GPU | NVIDIA RTX A5500 24GB ×2 | 支持DIA-NN分析中等规模数据(n<100),24GB显存满足大多数靶向分析需求 |
| 内存 | 256GB DDR5-5600 ECC(4通道) | 磷酸化蛋白质组学(富集后复杂度增加10倍)分析需 200GB+内存 支持MaxQuant-phospho |
| 存储 | 2TB NVMe Gen5(系统)+ 8TB NVMe(热数据) | 快速加载Skyline文档(.sky.zip)与谱图库,支持实时峰提取 |
| 软件栈 | 预装Claude Scientific Skills + Skyline + MaxQuant | 容器化部署,支持Windows/Ubuntu双系统启动 |
四、最热门应用场景与实战案例
4.1 临床大队列生物标志物发现(Precision Medicine)
技术路线:DIA采集(timsTOF Pro)→ DIA-NN深度学习鉴定 → DirectLFQ无标记定量 → MSstats统计检验 → 机器学习 biomarker panel构建(LASSO/RF)→ 临床验证(ELISA/PRM)
硬件需求:方案B(GX660M),重点配置 1TB内存 以支持 1000+样本 的蛋白质强度矩阵分析,4×A6000 加速DIA-NN深度谱图匹配。
4.2 单细胞蛋白质组学(Single-Cell Proteomics)
技术路线:SCoPE-MS/Drop-it采集 → DIA-NN-singlecell算法 → 肽段级批次效应校正(HarmonizR)→ 细胞聚类(Seurat-like分析)→ 差异轨迹分析(Slingshot)→ 细胞通讯(CellPhoneDB)
硬件需求:方案A(GT430M),512GB内存 支持单细胞特定谱图库(SCoPE-Lib)加载,双A6000 加速超低丰度肽段的AI识别。
4.3 4D-蛋白质组学与深度覆盖(Deep Proteome)
技术路线:timsTOF Pro 4D采集(PASEF)→ 离子淌度对齐 → DIA-NN 4D算法 → 深度分馏(24-fraction)整合 → 蛋白质互作网络(STRING)
硬件需求:方案B(GX660M),NVMe RAID 支撑4D数据(~20GB/样本)的高速读取,多GPU 并行处理多个分馏组分。
4.4 翻译后修饰(PTM)组学分析(Phospho/Acetyl/Ubiquityl)
技术路线:TiO2/IMAC富集 → DDA采集 → MaxQuant-PTM算法 → AI辅助位点定位(pDeep)→ 激酶底物预测(Kinase-Substrate Enrichment Analysis)
硬件需求:方案C(EX660),256GB内存 支持修饰肽段搜索空间扩大(考虑多种修饰组合),AVX-512 加速位点定位算法。
五、优化建议与最佳实践
-
GPU内存管理:
-
DIA-NN使用
--use-gpu参数时,设置--temp指向NVMe SSD(而非系统盘),临时文件可达 100GB+ -
Prosit预测时采用 Batch Size = 4096 以最大化A6000显存利用率(48GB可支持该批次)
-
-
存储I/O优化:
-
将质谱原始数据(.raw/.d)转换为 mzML 格式并压缩(zlib),可减少 50% 存储占用且加速随机读取
-
使用 ThermoRawFileParser 或 Bruker TDF-SDK 进行并行转换,多核CPU(>32核)可显著提升转换速度
-
-
Claude Scientific Skills自动化:bash
# 在Claude Code中加载蛋白质组学技能 /plugin marketplace add K-Dense-AI/claude-scientific-skills /plugin install maxquant@claude-scientific-skills /plugin install diann@claude-scientific-skills # 自动化DIA分析流程示例 "使用DIA-NN分析100个DIA文件,物种为人,使用DIA-NN预训练模型,启用GPU加速,FDR设置为1%,最后输出蛋白质定量矩阵并进行差异表达分析" -
内存管理策略:
-
MaxQuant分析大队列时,设置
--max-ram-threads为物理内存的 80%,避免系统交换(Swapping)导致性能崩溃 -
对于 >1TB 的原始数据集,采用 分块处理(Chunk Processing) 策略,DIA-NN支持按RT窗口分块分析
-
结语
蛋白质组学正从 "大数据" 向 "智能数据" 演进。当 DIA-NN 的深度学习算法能从噪声中提取低丰度信号,当 Prosit 能预测任意肽段的谱图特征,传统的计算架构已成为限制生物学发现的关键瓶颈。基于 Claude Scientific Skills 框架构建的AI蛋白质组学平台,配合 UltraLAB 的大显存GPU、TB级内存、高速NVMe存储架构,可将大队列临床样本的分析周期从 数月 压缩至 数日,将单细胞蛋白质组的鉴定深度推向 >3,000种蛋白质 的新高度。
在精准医学与药物开发的竞争中,选择经过AI算法优化的蛋白质组学硬件平台,本质上是购买发现疾病生物标志物的概率——让等待数据库搜索完成的时间,转化为解析疾病机制的科学洞察。
参考文献与资源:
-
K-Dense AI. (2025). Claude Scientific Skills: A set of ready to use Agent Skills for research, science, engineering, analysis, finance and writing. GitHub Repository. https://github.com/K-Dense-AI/claude-scientific-skills
-
涵盖MaxQuant、DIA-NN、Prosit、AlphaPept、OpenMS等140+科学技能模块
UltraLAB定制图形工作站 专注高端科研计算20年
咨询电话 400-7056-800
微信号 xasun001
上一篇:没有了


